Beta-Thalassämie ist eine vererbte Anämie, bei der der Körper normalerweise kein Hämoglobin produzieren kann. Hämoglobin A (das größte Hämoglobin bei Erwachsenen) enthält zwei Alpha-Globinketten und zwei Beta-Globinketten. In der Beta-Thalassämie ist das Knochenmark nicht in der Lage, eine normale Menge an Beta-Globin zu produzieren, was zu wenig Hämoglobin A führt. Außerdem gibt es einen Überschuss an Alpha-Globin, das nicht an Beta-Globin gebunden ist, was einen Hämolyse-Ausbruch der roten Zellen verursacht.
Wer ist gefährdet?
Beta-Thalassämie ist eine erbliche Erkrankung. Es erfordert, dass beide Eltern Träger der Störung sind, die Beta-Thalassämie-Eigenschaft oder Minderheit genannt wird. Wenn beide Eltern Beta-Thalassämie-Eigenschaft haben, haben sie eine 1 zu 4 Chance, ein Kind mit Beta-Thalassämie-Krankheit zu haben. Leider, weil Beta-Thalassämie-Eigenschaft keine Symptome verursacht, können Eltern dieses Risiko vor der Schwangerschaft nicht bewusst sein.
Was sind die Arten von Beta-Thalassämie?
Beta-Thalassämie kann auf zwei Arten klassifiziert werden: durch die genetische Mutation ererbt oder durch Transfusion benötigt.
Genetische Mutationen
Beta-Null-Thalassämie The: Die Null in der β-Null-Mutation zeigt an, dass kein Beta-Globin von diesem Chromosom produziert werden kann.Beta plus Thalassämie
: Beta-Globin wird in reduzierten Mengen produziert. Die Menge an produziertem Beta-Globin variiert stark mit der speziellen vererbten Mutation. Einige Mutationen produzieren fast kein Beta-Globin, was sie klinisch ähnlich zu Beta-Null macht.Transfusionsbedarf
Beta-Thalassämie major:
Beta-Thalassämie major ist definiert durch die Notwendigkeit lebenslanger Transfusionen, in der Regel monatlich (als Hypertransfusionstherapie bezeichnet).Beta Thalassaemia intermedia Usually: In der Regel moderate Anämie, die gelegentlich Transfusionen (während Krankheiten, Pubertät, etc.) erfordern kann, aber nicht regelmäßig.
Beta-Thalassämie-Merkmal / geringes: Dies führt zu einer leichten asymptomatischen Anämie, die normalerweise bei routinemäßigem Blutbild auftritt.
Wie wird Beta-Thalassämie diagnostiziert?Im Allgemeinen wird Beta-Thalassämie major auf dem Neugeborenen-Screening identifiziert. Bei diesen Säuglingen wird der Test nur Hämoglobin F (oder fetal), kein Hämoglobin A identifizieren. Diese Kinder werden an einen Hämatologen überwiesen, der genau überwacht werden soll. Einige stärker betroffene Patienten mit Beta-Thalassämie-Intermedia werden ebenfalls auf diese Weise identifiziert.
Einige leichter betroffene Patienten werden auf Routine-Blutbild (CBC) identifiziert werden. Die CBC zeigt eine leichte bis mäßige Anämie mit sehr kleinen roten Blutkörperchen. Dies kann mit Eisenmangelanämie verwechselt werden. Die Diagnose wird durch ein Hämoglobinprofil (auch Elektrophorese genannt) bestätigt. In der Beta-Thalassämie Intermedia und Merkmal dieser Prüfung zeigt Erhöhung in Hämoglobin A2 (eine zweite Form von Erwachsenen Hämoglobin) und manchmal F (fetalen).
Was sind die Behandlungen für Beta-Thalassämie?
Beta-Thalassämie-Merkmal erfordert keine Behandlung. Menschen mit Beta-Thalassämie-Eigenschaft haben lebenslange leichte Anämie.
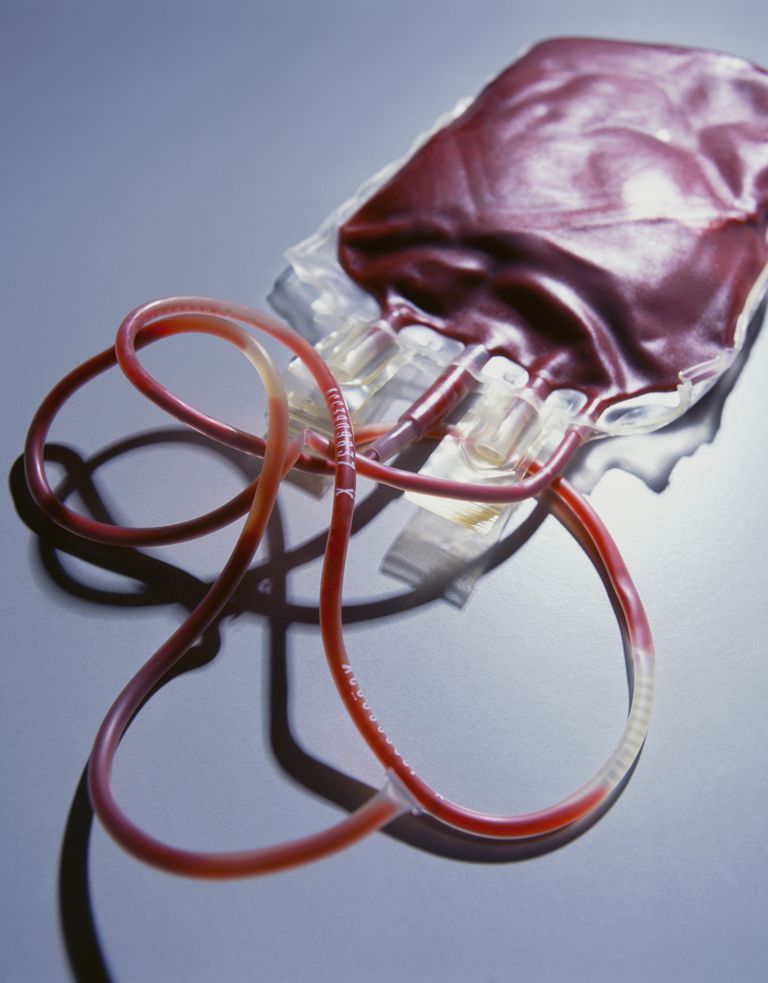
Transfusion:
Patienten mit Beta-Thalassämie major erfordern lebenslange Transfusionen. Die Transfusionstherapie dient zur Unterdrückung der Produktion roter Blutkörperchen im Knochenmark.
Patienten mit Beta-Thalassämie Intermedia können eine Transfusionstherapie bei Krankheiten und Pubertät (während Wachstumsschub) benötigen.Eisen-Chelat-Therapie:
Patienten mit Beta-Thalassämie major erhalten überschüssiges Eisen durch Erythrozytentransfusionen. Dieses überschüssige Eisen kann Organe, insbesondere das Herz schädigen. Eisenchelation unterstützt den Körper bei der Entfernung von Eisen. Diese können als orale Medikation oder als Infusion unter die Haut gegeben werden. Patienten mit Beta-Thalassaemia intermedia können auch bei fehlender Bluttransfusion eine Eisenüberladung entwickeln, die auf eine erhöhte Eisenresorption im Dünndarm zurückzuführen ist.
Splenektomie: Die Milz kann durch den Abbau von roten Blutkörperchen (Hämolyse) und die Produktion von roten Blutkörperchen in der Milz massiv vergrößert werden. Wenn die Transfusion erhöht werden muss oder andere Blutzellen aufgrund von Fallen in der Milz abfallen, muss die Milz chirurgisch entfernt werden.
Hydroxyharnstoff:Hydroxyharnstoff wurde verwendet, um die fetale Hämoglobinproduktion zu erhöhen. Der Erfolg war variabel.

