Übersicht
Phenylketonurie (Fen-ul-Keeton-YU-ree-ah oder PKU) ist eine erbliche Stoffwechselstörung, bei der der Körper das Protein (Aminosäure) Phenylalanin nicht vollständig abbauen kann. Dies geschieht, weil ein notwendiges Enzym, Phenylalaninhydroxylase, defizient ist. Aus diesem Grund baut sich Phenylalanin in den Körperzellen auf und verursacht Nervensystem- und Hirnschäden.
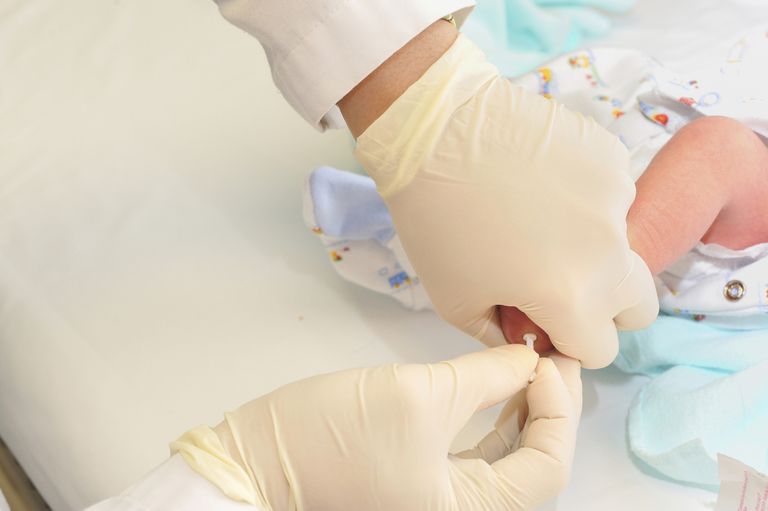
Phenylketonurie ist eine behandelbare Krankheit, die leicht durch einen einfachen Bluttest nachgewiesen werden kann. In den Vereinigten Staaten müssen alle Neugeborenen im Rahmen eines metabolischen und genetischen Screenings, das bei allen Neugeborenen durchgeführt wird, auf PKU getestet werden. Alle Neugeborenen in Großbritannien, Kanada, Australien, Neuseeland, Japan, den westlichen und den meisten osteuropäischen Ländern sowie vielen anderen Ländern auf der ganzen Welt werden ebenfalls getestet. Screen (Screening für PKU bei Frühgeborenen ist aus verschiedenen Gründen anders und schwieriger.)
Jedes Jahr werden 10.000 bis 15.000 Babys mit der Krankheit in den Vereinigten Staaten geboren und Phenylketonuria kommt bei Männern und Frauen aller ethnischen Herkunft vor (obwohl es ist häufiger bei Personen mit nordeuropäischem und indianischem Erbe.)
Symptome
Ein Säugling, der mit Phenylketonurie geboren wurde, entwickelt sich in den ersten Monaten normal.
Wenn sie unbehandelt bleiben, entwickeln sich die Symptome im Alter von 3 bis 6 Monaten und können einschließen: verzögerte Entwicklung mentale Retardierung
- Krampfanfälle
- sehr trockene Haut, Ekzem und Hautausschläge
- ausgeprägter mousy oder muffiger Geruch der Urin, Atem und Schweiß
- heller Teint, helles oder blondes Haar
- Reizbarkeit, Unruhe, Hyperaktivität
- Diagnose
- Phenylketonurie wird durch einen Bluttest diagnostiziert, meist im Rahmen der routinemäßigen Screeningtests bei Neugeborenen innerhalb der ersten Lebenstage .
Wenn PKU vorhanden ist, wird der Phenylalaninspiegel im Blut höher als normal sein.
Der Test ist sehr genau, wenn er durchgeführt wird, wenn der Säugling mehr als 24 Stunden alt, aber weniger als sieben Tage alt ist. Wenn ein Säugling weniger als 24 Stunden alt getestet wird, wird empfohlen, dass der Test wiederholt wird, wenn das Kind eine Woche alt ist. Wie oben erwähnt, mussten Frühgeborene aus verschiedenen Gründen auf eine andere Art und Weise getestet werden, einschließlich einer Verzögerung der Fütterungen.
Behandlung
Da Phenylketonurie ein Problem beim Abbau von Phenylalanin ist, erhält der Säugling eine spezielle Diät, die extrem arm an Phenylalanin ist.
Zunächst wird eine spezielle low-Phenylalanin-Säuglingsnahrung (Lofenalac) verwendet.
Wenn das Kind älter wird, werden niedrige Phenylalanin-Nahrungsmittel in die Nahrung aufgenommen, aber keine proteinreichen Nahrungsmittel wie Milch, Eier, Fleisch oder Fisch sind erlaubt. Der künstliche Süßstoff Aspartam (NutraSweet, Equal) enthält Phenylalanin, daher werden Diätgetränke und Lebensmittel, die Aspartam enthalten, ebenfalls vermieden. Sie haben wahrscheinlich die Position auf Softdrinks, wie zum Beispiel Diät Cola, die darauf hinweisen, dass das Produkt nicht von Menschen mit PKU verwendet werden sollte.
Personen müssen während der Kindheit und Jugend auf einer Phenylalanin-eingeschränkten Diät bleiben.
Einige Personen sind in der Lage, ihre Diätbeschränkungen zu reduzieren, wenn sie älter werden.
Regelmäßige Bluttests sind erforderlich, um die Phenylalaninspiegel zu messen, und die Ernährung muss möglicherweise angepasst werden, wenn die Werte zu hoch sind. Zusätzlich zu einer eingeschränkten Diät können einige Personen das Medikament Kuvan (Sapropterin) einnehmen, um den Phenylalaninspiegel im Blut zu senken.
Monitoring
Wie bereits erwähnt, Bluttests werden verwendet, um Menschen mit PKU zu überwachen. Derzeit empfehlen die Leitlinien, dass die Zielblutkonzentration von Phenylalanin bei Personen mit PKU aller Altersgruppen zwischen 120 und 360 μM liegen sollte. Manchmal ist ein Limit von bis zu 600 μM für ältere Erwachsene erlaubt. Schwangere Frauen müssen jedoch ihre Diät strenger befolgen, und ein Höchstwert von 240 μM wird empfohlen.
Studien, die sich mit Compliance befassen (die Anzahl der Menschen, die ihre Ernährung einhalten und diese Richtlinien einhalten), liegen bei Kindern zwischen der Geburt und dem vierten Lebensjahr bei 88 Prozent, bei Personen ab 30 Jahren jedoch nur bei 33 Prozent.
Rolle der Genetik
PKU ist eine genetische Störung, die von Eltern an Kinder weitergegeben wird. Um PKU zu haben, muss ein Baby eine spezifische Genmutation für PKU von jedem Elternteil erben. Wenn das Baby das Gen nur von einem Elternteil erbt, trägt das Baby auch die Genmutation für PKU, hat aber keine PKU.
Diejenigen, die nur eine Genmutation erben, entwickeln keine PKU, können aber die Bedingung an ihre Kinder weitergeben (Träger sein). Wenn zwei Eltern das Gen tragen, haben sie ungefähr 25 Prozent Chance, ein Baby mit PKU zu bekommen, a Eine 25-prozentige Chance, dass ihr Kind keine PKU entwickelt oder Trägerin ist, und eine 50-prozentige Chance, dass ihr Kind auch Träger der Krankheit sein wird.
Sobald PKU bei einem Baby diagnostiziert wird, muss dieses Baby während seines gesamten Lebens einem PKU-Ernährungsplan folgen. PKU in der Schwangerschaft Junge Frauen mit Phenylketonurie, die keine Phenylalanin-eingeschränkte Diät zu sich nehmen, haben hohe Phenylalaninwerte, wenn sie schwanger werden. Dies kann zu ernsthaften medizinischen Problemen führen, die als PKU-Syndrom für das Kind bekannt sind, einschließlich geistiger Behinderung, niedrigem Geburtsgewicht, Geburtsfehlern im Herzen oder anderen Geburtsfehlern. Wenn die junge Frau jedoch mindestens 3 Monate vor der Schwangerschaft eine Phenylalanin-Diät einnimmt und während der Schwangerschaft weiter ernährt, kann das PKU-Syndrom verhindert werden. Mit anderen Worten, eine gesunde Schwangerschaft ist für Frauen mit PKU möglich, solange sie planen und ihre Ernährung während der Schwangerschaft sorgfältig überwachen.
Forschung
Forscher suchen nach Möglichkeiten, die Phenylketonurie zu korrigieren, z. B. indem sie das defekte Gen, das für die Störung verantwortlich ist, ersetzen oder ein genetisch manipuliertes Enzym schaffen, das das defiziente ersetzt. Wissenschaftler untersuchen auch chemische Verbindungen wie Tetrahydrobiopterin (BH4) und große neutrale Aminosäuren als Wege zur Behandlung von PKU durch Senkung des Phenylalaninspiegels im Blut.
Bewältigung
Der Umgang mit PKU ist schwierig und erfordert großes Engagement, da es ein lebenslanges Unterfangen ist. Unterstützung kann hilfreich sein und es gibt viele Unterstützungsgruppen und Unterstützungsgemeinschaften, in denen Menschen mit anderen Personen, die mit PKU umgehen, interagieren können, sowohl für emotionale Unterstützung als auch um auf dem neuesten Stand der Forschung zu bleiben.
