Ohne Neugeborenenscreening wird Mukoviszidose (CF) erst diagnostiziert, wenn Symptome auftreten, und sind so weit fortgeschritten, dass Wachstumsstörungen und Atemprobleme auftreten. Üblicherweise geschieht dies in den ersten zwei Lebensjahren – aber zu dem Zeitpunkt, zu dem die Diagnose gestellt wird, ist der Schaden bereits aufgetreten.
Mit Neugeborenen-Screening wird jedoch CF in den ersten Wochen des Lebens diagnostiziert und Babys können sofort mit der Behandlung beginnen, wodurch die Unterernährung und Atemnot vermieden werden, die sonst unvermeidlich wären.
Eine frühzeitige Diagnose und Behandlung kann auch die Lebenserwartung von Menschen mit CF erhöhen. Menschen, die derzeit mit Mukoviszidose leben, können realistischerweise damit rechnen, bis Mitte dreißig zu leben, aber einige Studien sagen voraus, dass Babys, die heute mit Mukoviszidose geboren werden, aufgrund ihrer frühen Diagnose und besseren Behandlung in die 50er Jahre leben werden.
Screening-Prozess
Neugeborenen-Screening für zystische Fibrose ist ein Test, um die Möglichkeit having der Mukoviszidose zu erkennen. Es ist der erste Schritt in einem Prozess, der letztlich zu einer Diagnose von Mukoviszidose, Identifizierung von CF-Trägern führt oder beide Möglichkeiten eliminiert. Wenn die Ergebnisse des ersten Screenings positiv sind, bedeutet das nicht, dass ein Baby Mukoviszidose hat. Ein positives Screening bedeutet lediglich, dass weitere Tests durchgeführt werden müssen, um die Signifikanz des positiven Ergebnisses zu bestimmen. Erster Schritt – Erhöhung der roten Flagge
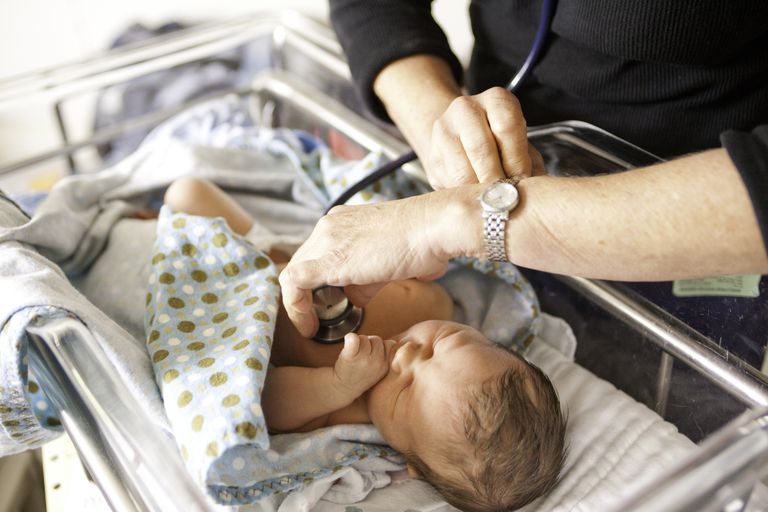
Der erste Schritt beim Neugeborenen-Screening ist ein Bluttest, der einige Tage nach der Geburt durchgeführt wird.
Blut wird aus dem Baby entnommen und in ein staatliches Labor geschickt, um nach verschiedenen Störungen zu suchen. Der Screening-Test für CF sucht nach erhöhten Spiegeln einer Substanz namens immunoreaktives Trypsinogen (IRT), einem Enzym, das von der Bauchspeicheldrüse gebildet wird. Babys, die mit Mukoviszidose geboren werden, haben oft einen hohen IRT-Spiegel im Blut, aber andere Bedingungen können auch dazu führen, dass das Enzym erhöht wird.
Wenn ein Neugeborenen-Screening auf CF positiv ist, bedeutet dies, dass hohe IRT-Werte festgestellt wurden. Jeder Staat legt seine eigenen Richtlinien für normale und abnormale IRT-Niveaus fest. Einige Staaten haben einen festen Wert, der ein positives Niveau darstellt, und einige Staaten wählen einen Prozentsatz der höchsten IRT-Werte aus, die jeden Tag gemeldet werden.
Zweiter Schritt – Gentests
Wenn das erste Neugeborenenscreening für IRT positiv ist, führen die meisten Staaten einen weiteren Test auf das Blut durch, um herauszufinden, ob das Baby eine Mutation des Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) hat. Es gibt mehr als 1.200 bekannte Mutationen im CFTR-Gen, die zystische Fibrose verursachen. Es ist nicht praktisch oder finanziell durchführbar, für alle von ihnen zu testen, aber die meisten Staaten testen auf einige der häufigsten Mutationen. Auch hier entscheidet jeder Staat, welche Mutationen in das Testpanel aufgenommen werden sollen.
Wenn eine Mutation des CFTR-Gens entdeckt wird, ist das Baby entweder ein CF-Träger oder hat eine Mukoviszidose-Erkrankung. Das staatliche Labor informiert den Hausarzt des Babys, und in einigen Staaten werden sie auch die Gesundheitsabteilung des Bezirks oder eine andere Behörde informieren, die für die Nachsorge von Familien zugelassen und ausgebildet ist.
Wenn die IRT-Werte erhöht sind, aber keine Mutation des CFTR-Gens festgestellt wird, werden die Ergebnisse beider Tests an den Hausarzt des Babys gesendet, der feststellen wird, ob weitere Tests erforderlich sind.
Dritter Schritt – Schweißtest
Der Schweisschloridtest, oder Schweißtest, ist seit vielen Jahren der Goldstandardtest zur Diagnose von Mukoviszidose. Der Test misst die Menge an Salz im Schweiß einer Person, die bei Menschen mit CF höher ist als normal. Ein Chloridgehalt von mehr als 60 mmol / Liter wird als positives Ergebnis angesehen.
Wenn der Hausarzt die Ergebnisse des Neugeborenen-Screenings vom staatlichen Labor erhält, entscheidet er oder sie, ob weitere Tests durchgeführt werden sollen. Wenn die IRT-Werte erhöht waren, aber keine CFTR-Mutation festgestellt wurde, kann der Hausarzt ohnehin einen Schweißtest anordnen, nur wenn das Baby eine der weniger häufigen Mutationen hat, die nicht in das genetische Testpanel aufgenommen wurde.
Wenn das Neugeborenenscreening eine CFTR-Mutation entdeckt, wird der Hausarzt einen Schweißtest anordnen, um festzustellen, ob das Baby eine CF-Erkrankung hat. Wenn der Schweißchlorid-Test positiv ist, hat das Baby eine zystische Fibrose und wird normalerweise an ein akkreditiertes CF-Zentrum überwiesen, um mit der Behandlung zu beginnen. Wenn genetische Tests eine Mutation im CFTR-Gen zeigten, der Schweißchlorid-Test jedoch negativ ist, ist das Baby ein CF-Träger, hat jedoch keine Krankheit und benötigt keine Behandlung. In jedem Fall wird in der Regel eine genetische Beratung mit der Familie durchgeführt, um die langfristigen Auswirkungen der Ergebnisse zu erklären.

