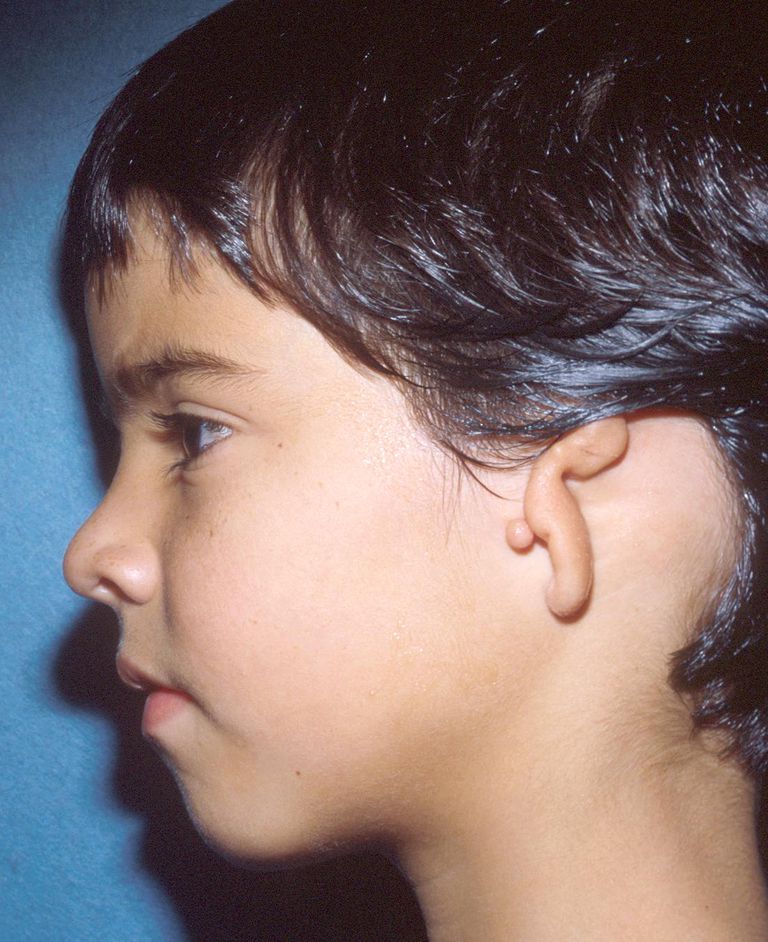
Mikrotie ist eine Ohrdeformität, die bei einer von 6.000 bis 12.000 Geburten auftritt. Es gibt mehrere Grade von Mikrotie, die von einem Teil des äußeren Ohres bis zum vollständigen Fehlen eines äußeren Ohres reichen. Wenn eine Mikrotie vorhanden ist, ist normalerweise kein Gehörgang vorhanden. Dies nennt man Atresie.
Ein Kind mit Mikrotie und Atresie hat normalerweise ein normales sensorineurales Gehör und einen mittleren bis schweren Schallleitungsschwerhörigkeit.
Das bedeutet, dass das Innenohr des Kindes normal ist (normaler sensorineuraler Status), aber da kein Gehörgang vorhanden ist, kann der Schall nicht über den Gehörgang zum Innenohr "geleitet" werden. Wenn ein Hörverlust aufgrund eines Außen- oder Mittelohrproblems auftritt, wird dies als Schallleitungsschwerhörigkeit bezeichnet. Wenn ein Hörverlust aufgrund eines Innenohrproblems auftritt, wird dies als sensorineuraler Hörverlust bezeichnet.
Es wird angenommen, dass Mikrotie durch Ischämie oder verminderte Durchblutung während der Entwicklung verursacht werden kann.
Zum Glück bildet sich das Innenohr (der Ort der Hör- und Gleichgewichtsorgane) zu einem anderen Zeitpunkt als das Außen- und Mittelohr. Dadurch funktioniert das Innenohr auch bei einem Kind mit Mikrotie normal. Die Nieren entwickeln sich ungefähr zur gleichen Zeit wie die Ohren, daher wird eine Bewertung üblicherweise vor dem Verlassen des Krankenhauses nach der Geburt durchgeführt. Microtie ist häufiger bei japanischen und Navaho Indianern, und auch häufiger bei Männern.
Wenn nur ein Ohr betroffen ist, ist typischerweise das rechte Ohr betroffen. Etwa 10% der Zeit sind beide Ohren beteiligt. Die Wahrscheinlichkeit, dass diese Erkrankung in einer zukünftigen Schwangerschaft wieder auftritt, beträgt weniger als 6%.
Die erste Priorität besteht darin, das Gehör des Kindes zu testen. In allen Fällen ist es wichtig, dem Kind einen maximalen Klang zu geben, um die Entwicklung des Gehirns und die Sprachentwicklung zu maximieren.
Ein BAER-Test (Gehirn-auditorische evozierte Reaktion) wird empfohlen, mit einem Verhaltens-Hörtest, wenn das Baby etwas älter ist. Ein CT-Scan (Computertomographie) wird ebenfalls durchgeführt, um die Anatomie der knöchernen Strukturen und der mittleren und inneren Ohren zu bestimmen.
Eltern mit Mikrotie neigen dazu, Schuldgefühle über den Zustand ihres Kindes zu haben. Es ist nicht die Schuld der Eltern!
Es ist wichtig, einen Genetikspezialisten zu sehen, um die Familiengeschichte zu bewerten. Einige Kinder mit Mikrotie haben andere Zustände wie das Goldenhar-Syndrom, das Treacher-Collins-Syndrom und die hemifaziale Mikrosomie (Unterentwicklung der Strukturen auf einer Seite des Gesichts). Kinder können auch Mittelohranomalien haben. Zahn- und Kieferanomalien können auch mit Mikrotie assoziiert sein.
Es gibt verschiedene Möglichkeiten, den Hörstatus von Kindern mit Mikro- und Atresie zu verbessern.
Ein knochenverankertes Hörgerät (BAHA) ist eine Option zur Gehörgangschirurgie zur Verbesserung des Gehörs.
Behandlungspläne werden aufgrund der unterschiedlichen Bedingungen für jedes Kind einzigartig sein. Ein interdisziplinäres Team muss zusammenarbeiten, um dem Kind eine optimal koordinierte Betreuung zu ermöglichen. Dieses Team besteht aus einem Hals-Nasen-Ohren-Arzt (Hals-Nasen-Ohren-Spezialist), einem plastischen Chirurgen, einem Audiologen (Hörspezialisten), einem genetischen Spezialisten, einem Zahnarzt, einem Logopäden und einem Kinderarzt oder Kinderkrankenschwester.
In einigen Fällen kann ein Sozialarbeiter oder Psychologe helfen, die Familie bei der Bewältigung dieser Krankheit zu unterstützen und Entscheidungen zu treffen.
Auch wenn ein Ohr nicht betroffen ist und gut hört, wird ein Knochenleitungshörgerät empfohlen, um eine auditorische Stimulation zu ermöglichen und die Entwicklung des auditorischen Gehirns auf der betroffenen Seite zu fördern. Bis das Kind für eine Operation alt genug ist, kann ein Knochenleitungshörgerät an einem stirnbandartigen Gurt getragen werden.
Videoclip: 6 Monate alter Junge mit rechtsseitiger Mikrotie und Atresie, der zum ersten Mal einen klaren Ton in seinem rechten Ohr hört. Die Veränderung in seinem Verhalten zeigt die Sekunde, dass er anfängt, klaren Ton in seinem rechten Ohr zu hören.
In der Regel wird das Kind etwa 4-10 Jahre alt sein, wenn eine Ohrrekonstruktion durchgeführt wird. Das äußere Ohr wird typischerweise rekonstruiert, manchmal unter Verwendung eines Stückes von Rippenknochen, um die Struktur des Ohres zu bilden. Dieses externe Ohr kann mehrere Monate vor der Operation der zweiten Stufe heilen, um die Gehörgänge zu schaffen.
Wenn Kinder auch Mittelohrdefekte (wie das Treacher-Collins-Syndrom) haben, kann die rekonstruktive Chirurgie des Gehörgangs das Gehör nicht verbessern. Eine Option kann ein knochenverankertes Hörgerät sein, das ideal für ein Kind ist, das Mittelohrprobleme hat, aber eine intakte Innenohr- und Nervenstruktur für das Gehirn.