Wenn Krebs die Knochen befällt, ist dies am häufigsten auf einen Nicht-Knochenkrebs zurückzuführen, der an anderer Stelle im Körper begonnen hat und sich auf die Knochen ausbreitet oder metastasiert. Im Gegensatz dazu liegt der Fokus hier auf den Krebsarten, die in den Knochen beginnen, die auch als primärer Knochenkrebs bekannt sind.
Primäre Knochenkrebs ist eigentlich eine breite Kategorie, bestehend aus vielen verschiedenen Arten von malignen Erkrankungen, von denen einige sehr selten sind; Von diesen sind jedoch Osteosarkom, Chondrosarkom und Ewing-Sarkom am häufigsten.
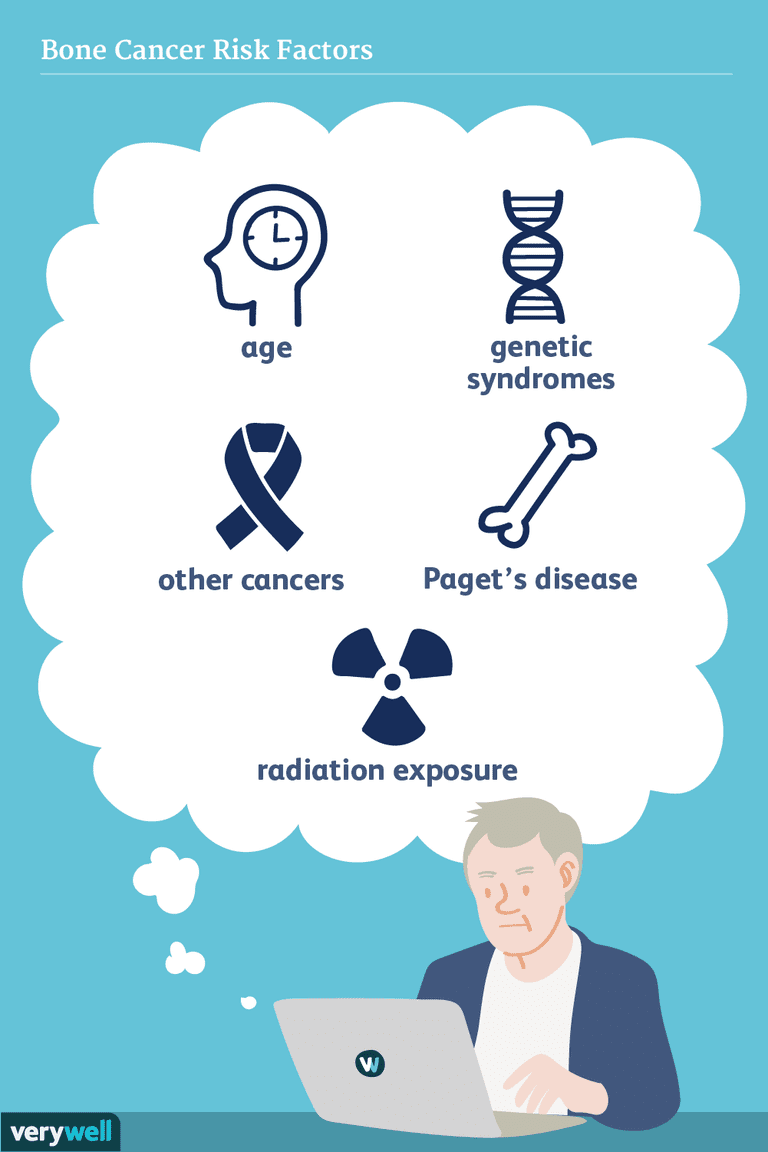
Bekannte Ursachen
Obwohl die Ursachen von Knochenkrebs nicht genau bekannt sind, ist bekannt, dass Veränderungen in der DNA der Krebszellen wichtig sind. In den meisten Fällen geschehen diese Veränderungen zufällig und werden nicht von Eltern an Kinder weitergegeben.
Wissenschaftler haben die Entwicklungsmuster untersucht, um zu versuchen, die beteiligten Risikofaktoren zu verstehen. Das Osteosarkom ist die dritthäufigste Form der Malignität, die Knochen bei Jugendlichen betrifft, der nur Leukämie und Lymphom vorausgehen. Chondrosarkom ist auch ein häufiger primärer Knochenkrebs, aber es ist häufiger bei Erwachsenen als bei Kindern und Jugendlichen, mit einem Durchschnittsalter bei der Diagnose von 51 Jahren. Ewing Sarkom wird am häufigsten bei Jugendlichen diagnostiziert, und das Durchschnittsalter der Diagnose beträgt 15 Jahre.
Osteosarkom-Risikoprofil
Osteosarkom ist der häufigste primäre Knochenkrebs insgesamt. Es gibt ein paar spezifische Bedingungen, von denen bekannt ist, dass sie die Chancen erhöhen, sie zu entwickeln. Menschen, die einen seltenen Tumor des Auges haben, bekannt als hereditäres Retinoblastom, haben ein erhöhtes Risiko, ein Osteosarkom zu entwickeln.
Darüber hinaus haben diejenigen, die zuvor mit Strahlentherapie und Chemotherapie gegen Krebs behandelt wurden, ein erhöhtes Risiko, später im Leben ein Osteosarkom zu entwickeln.
Übrigens sind sich die meisten Ärzte einig, dass gebrochene und verletzte Knochen und Sportverletzungen kein Osteosarkom verursachen. Solche Verletzungen können jedoch ein bereits bestehendes Osteosarkom oder einen anderen Knochentumor in ärztliche Behandlung bringen, so dass es definitiv eine Verbindung zwischen beiden gibt – es ist nur so, dass die mechanische Verletzung das Osteosarkom nicht zu verursachen scheint.
Alters-, Geschlechts- und ethnizitätsbezogene Risikofaktoren
Osteosarkom betrifft hauptsächlich zwei Hauptaltersgruppen – der erste Höhepunkt ist in den Teenager-Jahren und der zweite ist bei älteren Erwachsenen.
- Bei älteren Patienten entsteht das Osteosarkom in der Regel durch abnormale Knochen, z. B. bei Patienten mit langanhaltender Knochenkrankheit (z. B. Paget-Krankheit).
- Bei jungen Menschen ist das Osteosarkom vor dem fünften Lebensjahr extrem selten, und die Peak -Inzidenz tritt tatsächlich während des jugendlichen Wachstumsschubs auf. Ein repräsentatives "Standardalter" für Osteosarkom in der jüngeren Bevölkerung beträgt im Durchschnitt 16 Jahre für Mädchen und 18 Jahre für Jungen.
Osteosarkom ist relativ selten im Vergleich zu anderen Krebsarten; Es wird geschätzt, dass nur etwa 400 Personen jünger als 20 Jahre jedes Jahr in den Vereinigten Staaten mit Osteosarkom diagnostiziert werden. Jungen sind in den meisten Studien häufiger betroffen, und die Häufigkeit bei Jugendlichen afrikanischer Herkunft ist etwas höher als bei Weißen.
Risikofaktoren für jüngere Menschen
- Vorhandensein bestimmter seltener genetischer Krebssyndrome
- Alter zwischen 10 und 30 Jahren
- Hohe Körpergröße
- Männliches Geschlecht
- Afroamerikanische Rasse
- Vorhandensein bestimmter Knochenerkrankungen
Risikofaktoren für ältere Menschen
Bestimmte Knochenerkrankungen wie Morbus Paget, vor allem im Laufe der Zeit, sind mit einem erhöhten Risiko für Osteosarkom verbunden.
Dennoch ist das absolute Risiko gering: Nur etwa ein Prozent der Patienten mit Paget-Syndrom entwickelt jemals ein Osteosarkom.
Die Strahlenexposition ist ein gut dokumentierter Risikofaktor, und da der Abstand zwischen der Bestrahlung von Krebs und dem Auftreten von Osteosarkomen typischerweise länger ist (z. B. 10 Jahre oder mehr), ist dies für ältere Altersgruppen oft am relevantesten.
Genetische Prädispositionen
Prädisponierende genetische Syndrome für Osteosarkom umfassen:
- Bloom-Syndrom
- Diamond-Blackfan-Anämie
- Li-Fraumeni-Syndrom
- Morbus Paget
- Retinoblastom
- Rothmund-Thomson-Syndrom (auch poikiloderma congenitale genannt)
- Werner-Syndrom
- Der Funktionsverlust der Tumorsuppressorgene p53 und Retinoblastom spielt eine wichtige Rolle in der Osteosarkomentwicklung.
Obwohl Keimbahn (Ei und Sperma) Mutationen von p53 und Retinoblastom-Genen selten sind, sind diese Gene in der Mehrheit der Osteosarkom-Tumorproben verändert, so dass es einen Zusammenhang mit der Entwicklung von Osteosarkom gibt. Keimbahnmutationen im p53-Gen können zu einem hohen Risiko für die Entstehung von Malignomen führen, einschließlich des Osteosarkoms, das als Li-Fraumeni-Syndrom beschrieben wurde.
Obwohl Veränderungen in Tumorsuppressorgenen und Onkogenen notwendig sind, um Osteosarkome zu erzeugen, ist nicht klar, welches dieser Ereignisse zuerst auftritt und warum oder wie es auftritt.
Osteosarkome bei Patienten mit Morbus Paget
Es gibt eine seltene Untergruppe von Osteosarkomen, die eine sehr schlechte Prognose haben. Die Tumoren treten meist bei Personen auf, die älter als 60 Jahre sind. Die Tumoren sind zum Zeitpunkt ihres Auftretens groß und neigen dazu, sehr destruktiv zu sein, was es schwierig macht, eine vollständige chirurgische Resektion (Entfernung) zu erhalten, und Lungenmetastasen sind häufig von Anfang an vorhanden.
Das Risikoprofil ist das einer älteren Altersgruppe. Sie entwickeln sich bei etwa einem Prozent der Menschen mit Morbus Paget, meist wenn viele Knochen betroffen sind. Die Tumore treten tendenziell im Hüftknochen, Oberschenkelknochen nahe der Hüfte und im Armknochen nahe dem Schultergelenk auf; Sie sind schwer operativ zu behandeln, hauptsächlich wegen des Alters des Patienten und der Größe des Tumors.
Manchmal ist eine Amputation notwendig, besonders wenn der Knochen aufgrund des Krebses bricht, was häufig vorkommt.
Parosteale und periostale Osteosarkome
Dies sind Teilgruppen, die aufgrund ihrer Lage im Knochen so genannt werden; sie sind normalerweise weniger aggressive Osteosarkome, die auf der Knochenoberfläche in Verbindung mit der Gewebeschicht, die den Knochen umgibt, oder dem Periost entstehen. Sie dringen selten in die inneren Teile des Knochens ein und werden selten zu hochmalignen Osteosarkomen.
Das Risikoprofil für das parosteale Osteosarkom unterscheidet sich von dem des klassischen Osteosarkoms: Es ist häufiger bei Frauen als bei Männern, am häufigsten in der Altersgruppe der 20- bis 40-Jährigen und tritt typischerweise in der hinteren Oberschenkelknochenregion nahe dem Knie auf Gelenk, obwohl jeder Knochen im Skelett betroffen sein kann.
Höhere Risiko-Prognose
Risikofaktoren wurden mit besseren und schlechteren Prognosen in Verbindung gebracht, aber leider waren diese Faktoren im Allgemeinen nicht hilfreich bei der Identifizierung von Patienten, die von intensiveren oder weniger intensiven therapeutischen Maßnahmen profitieren könnten, während sie hervorragende Ergebnisse erzielen. Zu den Faktoren, von denen bekannt ist, dass sie die Ergebnisse beeinflussen, gehören die folgenden.
Primäre Tumorstelle
Von Tumoren, die sich in den Armen und Beinen bilden, haben diejenigen, die weiter weg vom Körperkern oder Torso sind, eine bessere Prognose.
Primäre Tumoren, die sich im Schädel und in der Wirbelsäule bilden, sind mit dem größten Progressions- und Todesrisiko verbunden, hauptsächlich weil es schwieriger ist, an diesen Stellen eine vollständige chirurgische Entfernung des Krebses zu erreichen. Kopf- und Nacken-Osteosarkome im Kiefer- und Mundbereich haben eine bessere Prognose als andere primäre Stellen im Kopf- und Halsbereich, möglicherweise weil sie früher zur Aufmerksamkeit kommen.
Osteokarzinome aus dem Hüftknochen machen sieben bis neun Prozent aller Osteosarkome aus; Überlebensraten für Patienten sind 20 bis 47 Prozent.
Patienten mit multifokalem Osteosarkom (definiert als multiple Knochenläsionen ohne einen klaren Primärtumor) haben eine extrem schlechte Prognose.
Lokalisierte vs. metastasierte Erkrankung
Patienten mit lokalisierten Erkrankungen (keine Ausbreitung in entfernte Gebiete) haben eine viel bessere Prognose als Patienten mit metastasierten Erkrankungen. Nicht weniger als 20 Prozent der Patienten werden Metastasen auf Scans bei der Diagnose erkennen, wobei die Lunge die häufigste Stelle ist. Die Prognose für Patienten mit metastasierender Erkrankung scheint hauptsächlich durch Metastasen, Metastasenzahl und chirurgische Resektabilität der Metastasen bestimmt zu sein.
Bei Patienten mit metastasierten Erkrankungen scheint die Prognose besser zu sein, mit weniger Lungenmetastasen und wenn sich die Krankheit nur auf eine Lunge und nicht auf beide Lungen ausbreitet.
Tumor-Nekrose nach Chemotherapie
Tumor-Nekrose bezeichnet hier krebsartiges Gewebe, das durch die Behandlung "abgestorben" ist.
Nach Chemotherapie und Operation beurteilt der Pathologe die Tumornekrose im entfernten Tumor. Patienten mit mindestens 90 Prozent Nekrose im Primärtumor nach Chemotherapie haben eine bessere Prognose als Patienten mit weniger Nekrose.
Forscher stellen jedoch fest, dass weniger Nekrose nicht dahingehend interpretiert werden sollte, dass eine Chemotherapie unwirksam war; die Heilungsraten für Patienten mit geringer oder keiner Nekrose nach Induktions-Chemotherapie sind viel höher als die Heilungsraten für Patienten, die keine Chemotherapie erhalten.
Chondrosarkom Risikoprofil
Dies ist ein bösartiger Tumor von Knorpel produzierenden Zellen, und es stellt etwa 20 Prozent aller primären Knochentumoren. Ein Chondrosarkom kann allein oder sekundär in der sogenannten "malignen Degeneration" von gutartigen Tumoren (wie Osteochondrom oder benignes Enchondrom) auftreten. Risikofaktoren umfassen:
- Alter: tritt normalerweise bei Menschen über 40 Jahren auf; es tritt jedoch auch in der jüngeren Altersgruppe auf, und wenn es dies tut, tendiert es dazu, von einer höhergradigen Malignität zu sein, die zu Metastasen fähig ist.
- Geschlecht:Tritt bei beiden Geschlechtern nahezu gleich häufig auf.
- Lokalisation: Kann in jedem Knochen auftreten, aber es gibt eine Tendenz zur Entwicklung im Hüft- und Oberschenkelknochen. Chondrosarkom kann in anderen flachen Knochen, wie dem Schulterblatt, den Rippen und dem Schädel auftreten.
- Genetik: Das multiple Exostosesyndrom (manchmal als multiples Osteochondromsyndrom bezeichnet) ist eine erbliche Erkrankung, die viele Beulen an den Knochen einer Person verursacht, die hauptsächlich aus Knorpel bestehen. Exostosen können schmerzhaft sein und zu Knochenfehlbildungen und / oder Knochenbrüchen führen. Die Störung ist genetisch bedingt (verursacht durch eine Mutation in einem der 3 Gene EXT1, EXT2 oder EXT3), und Patienten mit dieser Erkrankung haben ein erhöhtes Risiko für Chondrosarkom.
- Andere gutartige Tumore: Ein Enchondrom ist ein gutartiger Knorpeltumor, der in den Knochen einwächst. Menschen, die viele dieser Tumoren bekommen, haben eine Multiple Enchondromatose. Sie haben auch ein erhöhtes Risiko, ein Chondrosarkom zu entwickeln.
Ewing Sarkom Risikoprofil
Diesesist viel häufiger unter Weißen (entweder nicht-hispanischen oder hispanischen) und weniger häufig unter den asiatischen Amerikanern und extrem selten unter den Afroamerikanern. Ewing Tumoren können in jedem Alter auftreten, aber sie sind am häufigsten bei Jugendlichen und sind weniger häufig bei jungen Erwachsenen und jungen Kindern. Sie sind selten bei älteren Erwachsenen.
Fast alle Ewing-Tumorzellen haben Veränderungen, die das EWS-Gen betreffen, das sich auf Chromosom 22 befindet. Die Aktivierung des EWS-Gens führt zum Überwachsen der Zellen und zur Entwicklung dieses Krebses, aber die genaue Art und Weise, wie dies geschieht, ist dies nicht noch klar.

